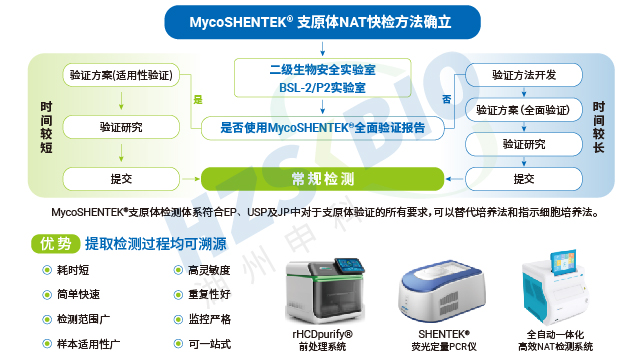
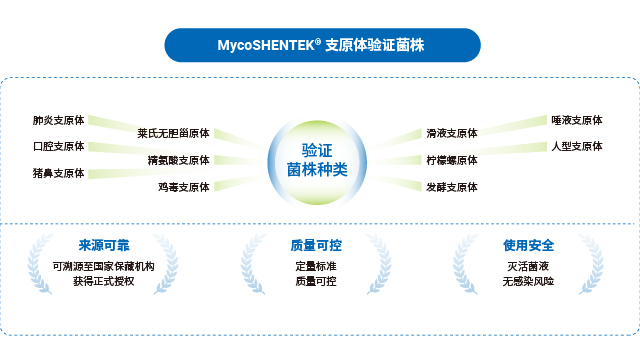
全球主流药典对支原体 NAT 检测的标准菌株选择形成了明确共识,中国、欧洲、美国、日本四国药典一致推荐优先使用猪鼻支原体、口腔支原体、肺炎支原体三种菌株,用于 NAT 方法检测限的验证,这一选择基于支原体的污染发生频率与进化关系。不同地区法规的验证范围略有差异:EP、USP、ChP 要求覆盖特异性、检测限、耐受性,WHO 还需包含半定量与定性实验;部分地区如欧洲药典额外纳入莱氏无胆甾原体、滑液支原体等菌株,针对昆虫和植物来源物料生产场景则需关注螺原体等特殊菌株。合规的标准菌株是 NAT 检测方法验证的前提,其溯源性与授权资质直接影响检测结果的认可度。
支原体检测 NAT 法引物设计需平衡覆盖范围与特异性,避免交叉反应。河南复杂基质支原体检测技术服务
湖州申科推出的 MycoSHENTEK® 支原体验证菌株,符合全球药典要求,目前已提供口腔支原体、肺炎支原体、猪鼻支原体三种菌株,后续将逐步扩充其他验证用菌株。该系列菌株溯源至全球保藏机构并获得正式授权,经培养法测定 CFU(菌落形成单位)与 dPCR 法测定 GC(基因组拷贝数),实现准确定量标定,浓度涵盖 10CFU 和 100CFU 两种规格,每盒含 3 管,完全满足支原体检测方法验证需求。菌株经灭活处理,无风险,使用时只需加入相应体积样品基质即可开展验证,操作便捷,同时通过双重定量检测确保质量可控,为 NAT 方法验证提供可靠的标准物质支撑。
浙江细胞疗法产品支原体检测快速检测支原体检测结果判定需结合 FAM 与 VIC 信号,警惕基质抑制导致的误判。
支原体检测中,污染引发的假阳性是生物制品企业的痛点。实验室环境控制不当、人员操作不规范、仪器耗材混用等因素,都可能导致核酸气溶胶污染或上样污染,进而影响检测结果。一旦出现假阳性,企业需花费大量时间排查验证,严重时会导致批次报废、影响其他产品正常生产。常规 NAT 检测法虽比传统培养法快速,但仍存在明显短板:需配备前处理设备、PCR 仪器等多重耗材,核酸提取、PCR 上机、高浓度产物处理等多个环节均有污染风险;实验准备需 40-60 分钟,操作耗时超 4 小时,每天只能完成 1-2 轮单一类型样本检测,且对场地和人员技能要求极高,需单独划分试剂准备区、样品制备区等多个区域,人力与场地成本高昂。
针对不同类型的样品基质,需采用定制化的支原体检测前处理方案,以消除干扰、提升检测效果。细胞悬液需经热处理、样品处理液作用、细胞碎片去除、浓缩离心,再 55℃消化;上清或高浓度质粒样品需先浓缩离心,再用样品处理液作用后 55℃消化;5% 人血白蛋白样品则采用浓缩离心 + 样品处理液作用 + 25℃消化的流程。常见干扰基质包括冻存保护剂、高浓度细胞、代谢产物等,优化前处理可遵循三大原则:提取前通过离心取上清或去除抑制剂预处理样品;高浓度蛋白样本提取时增加蛋白酶 K 用量,增强蛋白降解效果;细胞类样品适当降低细胞数或先裂解细胞,减少细胞基质对检测的干扰。
复杂样品在进行支原体检测时,可适当稀释样本或增加蛋白酶 K 用量,消除基质干扰。
选择合适的商业化支原体试剂盒需综合五大主要维度,确保检测合规、稳定与高效。一是监管认可度,需关注试剂盒验证的全面性与室间验证情况,菌株选择是否合规,以及厂家是否有丰富的客户申报案例;二是厂家资质,确认厂家是否接受供应商审计,能否提供 “产品 + 服务” 的全流程解决方案;三是产品设计,重点考察试剂盒方法学验证的严谨性、对复杂样品基质的耐用性与抗干扰能力,以及是否具备避免假阴 / 假阳性的质控设计,是否符合药典要求;四是技术支持,评估厂家的技术响应速度、问题解决能力,能否提供专业的验证指导;五是质量保障,关注试剂盒供应的稳定性与质量均一性,避免因产品批次差异影响检测结果,确保长期质控需求的稳定满足。
细胞疗法产品时效短,需用 核酸扩增NAT 法快速检测支原体,满足放行时效。浙江细胞疗法产品支原体检测快速检测
耐用性验证需确认方法参数微小变动时,支原体检测结果不受影响。河南复杂基质支原体检测技术服务
检测限验证是支原体 NAT 方法(核酸扩增法)合规性的关键要求,法规明确界定需为每种目标支原体确定阳性检测临界值。验证流程需满足严格的实验设计:每种支原体至少进行三次单独的 10 倍梯度稀释,每次稀释后需制备平行管检测,再确保各稀释浓度获得 24 个检测结果。阳性临界值的判定标准为,该浓度下至少 95% 的试验运行能得到阳性结果,即 24 个样品中需至少出现 23 个有效阳性结果。这一严谨设计旨在确保 NAT 检测方法在实际应用中,能够稳定检出低浓度支原体污染,避免因检测灵敏度不足导致的安全风险,为生物制品质量控制提供可靠保障。
河南复杂基质支原体检测技术服务