细菌内毒素是革兰氏阴性菌细胞壁外膜释放的脂多糖(LPS)成分,具有极强的生物活性,微量即可引发人体发热、休克甚至多脏器功能衰竭。因此,在生物制品、医疗器械、制药用水等领域,细菌内毒素检测是保障产品安全性的关键质控环节。其检测原理基于内毒素与特定试剂的特异性反应:内毒素可活化鲎血变形细胞裂解物(LAL)中的凝血级联反应,通过C因子通路触发酶促反应,通过观察凝胶形成、浊度变化或显色强度实现定量或定性分析。目前,各国药典均将内毒素检测列为强制要求,以降低临床应用中的热原风险。
细菌内毒素工作品若效价误差或不稳定,将影响实验结果。广东非动物源内毒素检测低内毒素回收
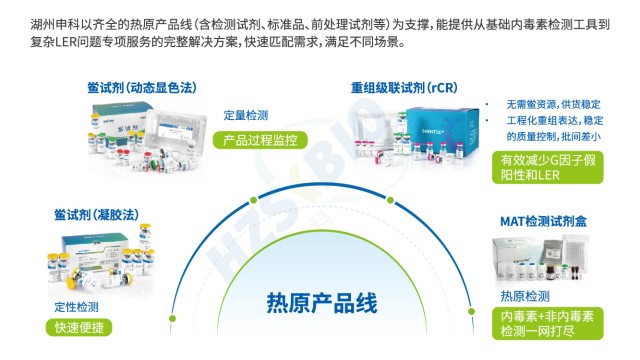
重组级联试剂(rCR)通过完整模拟天然鲎试剂的酶促级联反应路径,实现高效且特异的内毒素检测。其反应机制为:内毒素首先活化重组 C 因子,活化的 C 因子进一步活化重组 B 因子,随后活化重组凝固酶原转化为凝固酶,再催化显色底物产生黄色信号(405nm 波长可检测)。这一级联放大过程有效提升了检测灵敏度,即使微量内毒素也能产生可识别信号。与单因子的重组 C 因子法(rFC)相比,rCR 的多因子级联反应抗干扰能力更强,尤其对复杂基质样品(如高蛋白单抗、疫苗)表现更优。同时,rCR 剔除了天然鲎试剂中的 G 因子,避免了与 β-D 葡聚糖的非特异性反应,从根本上减少假阳性结果,保障检测数据的可靠性。
广东抗体药物内毒素检测法规要求含蛋白酶的样本致假阳性时,可稀释后 70℃加热 5-15min 灭活,再做内毒素检测。
动态显色法鲎试剂是湖州申科生物针对生产过程监控开发的内毒素检测工具,兼具准确性和便利性。该试剂灵敏度达 0.005-5EU/mL,标准曲线 R²≥0.990,可准确定量内毒素浓度,满足生物制品中间品和成品的放行需求。成套包装设计包含内毒素标准品、检查用水、主试剂复溶液、主反应试剂、96 孔板及封口膜,无需额外采购辅料,开箱即可使用,减少耗材浪费。稳定性上,批次间 CV 值≤15%,优于行业 20% 的平均水平,确保长期检测数据的一致性。配套抗增液可有效应对蛋白质、多糖等基质干扰,加标回收率稳定在 80%-120%。操作上适配主流酶标仪,支持 405nm 动态读数,数据可自动记录追溯,符合 GMP 数据完整性要求。
在为新物料或新产品、中间产品建立细菌内毒素检测方法时,常会遇到各种困难,尤其是尚处于新药研发早期阶段的药物。此时,由于药物制剂、缓冲系统等还不稳定,经常会发生变化,这样就给方法的建立带来了不同程度的影响。在建立细菌内毒素检查法之前,须尽可能多地了解有关该药品的基本信息,例如:有关样品的可溶性信息、推荐的稀释液、在水中的溶解度以及合适溶剂,样品的pH范围,分子量大小;如果是蛋白产品,还要了解该产品的等电点,产品规格、体积或重量,拟用于临床的用法和用量等,以便选择合适的样品处理方法和内毒素检测方法。
内毒素工作标准品(CSE)稀释液配制时涡旋很重要,可使内毒素充分溶解,保证浓度准确。
在进行内毒素检测时,干扰试验又叫增强或抑制试验,主要目的是确证检测内毒素的方法是否受样品干扰。在建立细菌内毒素检查方法中,验证试验前,要去除样品可能含有的内毒素,以确保建立方法的准确可靠。药典规定:①当进行新药的内毒素检查试验前,或无内毒素检查项品种建立内毒素检查法时,需进行干扰试验;②当鲎试剂、供试品的配方、生产工艺改变,或试验环境下发生了任何有可能影响试验结果的变化时,需重新进行干扰试验。生产厂家常发生的一些微小变更,会影响到评估结果,进而影响到供试品对鲎试剂的干扰试验。因此,生产厂家应制定一个重复进行干扰试验的周期,并进行跟踪和记录。
表面活性剂会改变内毒素活性,用重组级联试剂检测前需稀释样本,消除其对反应的干扰。
江苏非动物源内毒素检测外源性热原含细菌内毒素、脂磷壁酸、酵母多糖等,单核细胞活化反应检查法可检出全部热原。广东非动物源内毒素检测低内毒素回收
内毒素检测结果误差可能源于多环节:试剂方面,鲎试剂(LAL )或试剂批间差异、过期试剂活性下降会导致结果偏差,需通过试剂验收(如阳性对照回收率验证)确保质量;操作方面,实验器具未除热原(如玻璃器皿未干热灭菌)、加样体积不准确会引入污染或误差,需严格执行 SOP(如器皿 250℃干热灭菌≥30 分钟);环境方面,实验室空气中的微生物孢子、粉尘可能污染样品,需在洁净工作台操作并设置阴性对照。此外,反应温度波动(偏离 37℃±1℃)会影响酶活性,需使用恒温孵育器精确控温,确保反应条件稳定。
广东非动物源内毒素检测低内毒素回收