- 品牌
- 司鼎;OriCell
现在有些PCR因为扩增区很短,即使Taq酶活性不是很好也能在很短的时间内复制完成,因此可以改为两步法,即退火和延伸同时在60℃-65℃间进行,以减少一次升降温过程,提高了反应速度。检测:PCR反应扩增出了高的拷贝数,下一步检测就成了关键。荧光素(溴化乙锭,EB)染色凝胶电泳是很常用的检测手段。电泳法检测特异性是不太高的,因此引物两聚体等非特异性的杂交体很容易引起误判。但因为其简捷易行,成为了主流检测方法。近年来以荧光探针为的检测方法,有逐渐取代电泳法的趋势。在嵌套聚合酶链反应中,除了预期的靶之外,该产物可能仍然由非特异性扩增的DN段组成。武汉组织数字PCR研究方案
聚合酶链反应同时扩增单个精子中几个基因座的能力]增强了极大地增强了通过研究减数分裂后染色体交叉来进行基因定位的传统任务。通过分析数千个单个精子,已经直接观察到非常紧密基因座之间罕见的交叉事件。类似地,可以分析异常的缺失、插入、易位或倒位,所有这些都无需等待(或支付)漫长而艰苦的受精、胚胎发生等过程。定点突变:聚合酶链反应可用于产生突变基因,突变由科学家随意选择。可以选择这些突变来理解蛋白质是如何完成其功能的,并改变或改善蛋白质功能。杭州骨头Real-time PCR设计公司PCR的另一个限制是,即使是很少量的污染DNA也可以被扩增,导致误导或模糊的结果。

聚合酶链式反应的常见问题:Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特 异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。或100ul,应用多 大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul 后,再做大体积时,一定要模索条件,否则容易失败。物理原因:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。有时还有必要用标准的温度计,检测一下扩增仪或水溶锅内的变性、退火和延伸温度,这也是PCR失败的原因之一。
聚合酶链式反应的循环参数:预变性,模板DNA完全变性与PCR酶的完全对PCR能否成功至关重要,建议加热时间参考试剂说明书,一般未修饰的Taq酶时间为两分钟。变性步骤,循环中一般95℃,30秒足以使各种靶DNA序列完全变性,可能的情况下可缩短该步骤时间。变性时间过长损害酶活性,过短靶序列变性不彻底,易造成扩增失败。引物退火,退火温度需要从多方面去决定,一般根据引物的Tm值为参考,根据扩增的长度适当下调作为退火温度。然后在此次实验基础上做出预估。退火温度对PCR的特异性有较大影响。引物延伸,引物延伸一般在72℃进行(Taq酶很适温度)。但在扩增长度较短且退火温度较高时,本步骤可省略延伸时间随扩增片段长短而定,一般推荐在1000bp以上,含Pfu及其衍生物的衍生设定为1min/kbp。循环数,大多数PCR含25-35循环,过多易产生非特异扩增。延伸,在一个循环后,反应在72℃维持10-30分钟.使引物延伸完全,并使单链产物退火成双链。聚合酶链式反应中的DMSO、甘油等,主要用来保持酶的活性和帮助DNA解除缠绕结构。
聚合酶链反应:热启动聚合酶链式反应:一种在PCR的初始建立阶段减少非特异性扩增的技术。它可以通过将反应组分加热到变性温度(例如95 c)在加入聚合酶。[36]已经开发了特殊的酶系统,通过结合抗体来抑制聚合酶在环境温度下的活性[37][37]或者通过共价键结合的抑制剂的存在,该抑制剂在高温活化步骤后解离。热启动/冷完成聚合酶链反应是通过新的杂合聚合酶实现的,这些酶在环境温度下不活跃,在伸长温度下立即。序列间特异性聚合酶链反应(ISSR):一种用于DNA指纹识别的聚合酶链反应方法,它放大简单重复序列之间的区域,以产生扩增片段长度的独特指纹。电子聚合酶链反应(数字聚合酶链反应、虚拟聚合酶链反应、电子聚合酶链反应、电子聚合酶链反应)是指计算工具使用给定的一组引物 ( 探针)来扩增来自测序的基因组或转录组的DNA 序列,用于计算理论聚合酶链反应结果。电子PCR被提出作为分子生物学的教育工具。如果基因的基因组DNA序列是已知的,逆转录-聚合酶链反应可以用来绘制基因中外显子和内含子的位置。温州实时RT-PCR检测技术研究方案
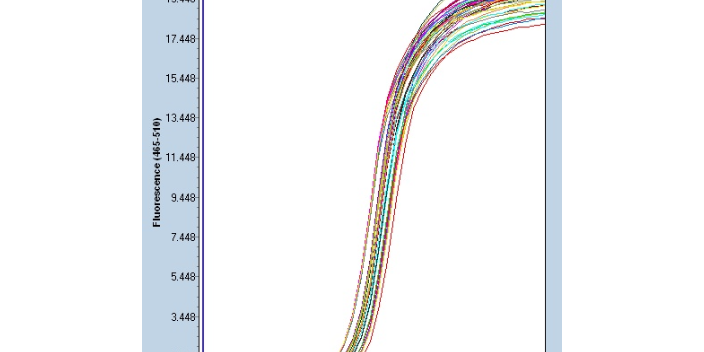
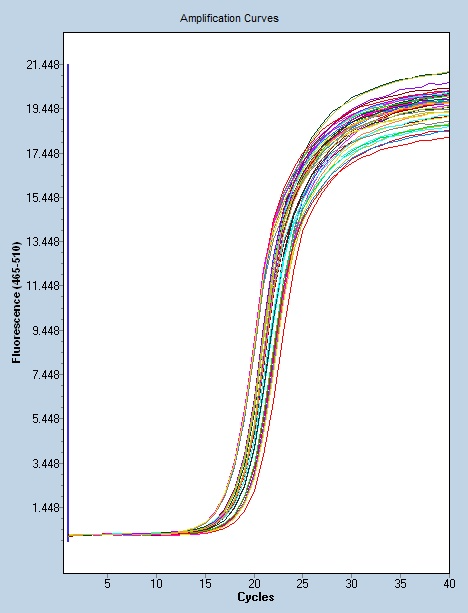
聚合酶链反应具有定量合成产物的优点。武汉组织数字PCR研究方案
聚合酶链式反应的常见问题:阴性:需注意的是有时忘加Taq酶或溴乙锭。引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称扩增,对策为:选定一个好的引物合成单位。引物的浓度不但要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应和引物合成单位协商解决。如一条引物亮度高,一条亮度低,在稀释引物时要平衡其浓度。引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质降解失效。引物设计不合理,如引物长度不够,引物之间形成二聚体等。武汉组织数字PCR研究方案

Real-time PCR-传统定量PCR:1)内参照法:在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板。2)竞争法:选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。PCR反应的很大特点是具有较大扩增能力与极高的灵敏性。深圳微量荧光PCR设计公司Real-time PCR原理:基于探针...
- 珠海实时荧光定量PCR技术服务 2023-08-11
- 温州组织荧光PCR原理及步骤 2023-08-11
- 上海微量RT-PCR检测技术供应商 2023-08-11
- 珠海骨头Real-time PCR原理 2023-08-10
- 武汉血液数字PCR技术服务 2023-08-10
- 连云港血液数字PCR原理及步骤 2023-08-10
- 宁波骨头定量PCR应用 2023-08-09
- 武汉血液PCR检测技术设计公司 2023-08-09
- 苏州细胞Real-time PCR服务 2023-08-09
- 徐州实时PCR检测技术服务 2023-08-09


- 杭州细胞Real-time PCR原理及步骤 2023-08-08
- 上海特殊样本数字PCR哪家好 2023-08-07
- 常州特殊样本PCR检测技术哪家好 2023-08-07
- 珠海特殊样本定量PCR原理 2023-08-07
- 徐州实时Real-time PCR供应商 2023-08-07
- 上海血液荧光定量PCR原理 2023-08-07



- 宁波动物细胞线性PEI转染试剂品牌 07-09
- 上海高效线性PEI转染试剂使用方法 07-09
- 无菌线性PEI转染试剂报价 07-09
- 南京HEK-293线性PEI转染试剂是干什么的 07-09
- 无锡高纯度线性PEI转染试剂作用 07-09
- 南京40000Da线性PEI转染试剂现货 07-08
- 武汉动物细胞线性PEI转染试剂品牌 07-08
- 上海细胞线性PEI转染试剂品牌 07-08
- 合肥血清兼容线性PEI转染试剂原理 07-08
- 细胞实验线性PEI转染试剂 07-08