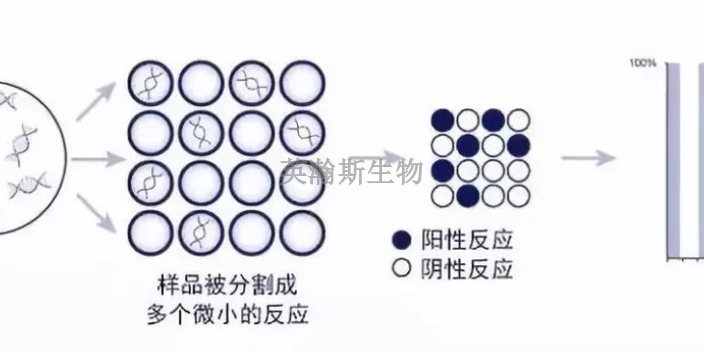
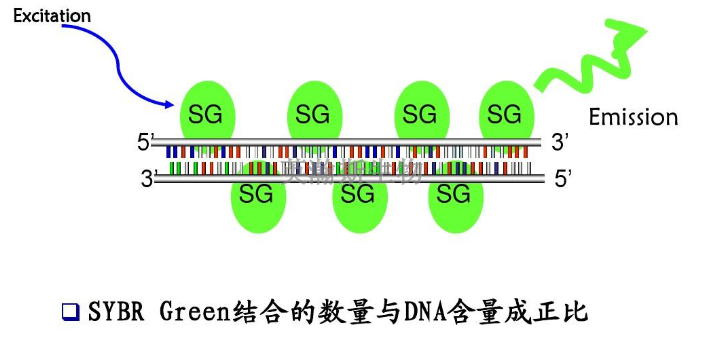
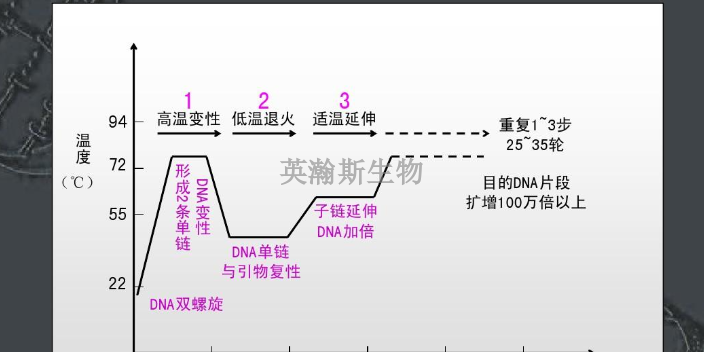
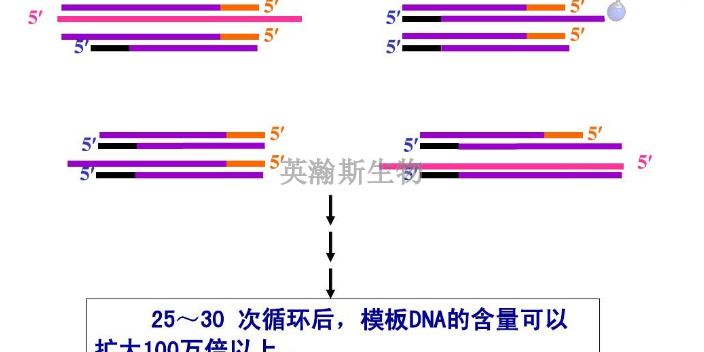
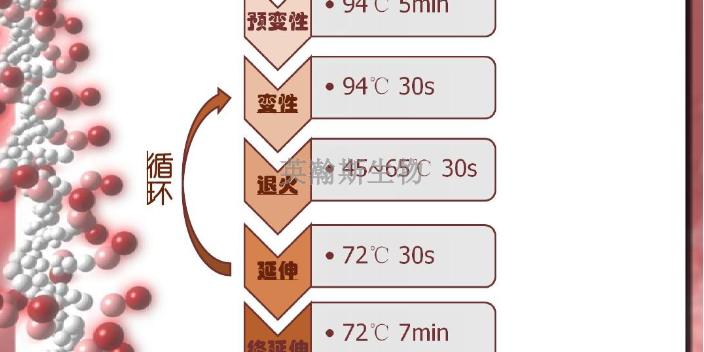
pcr是一种在体外扩增特定DNA序列的技术方法,通过与特定DNA区域两端互补的寡核苷酸为引物(primer)在试管中DNA聚合酶选择性地单独复制合成介于两引物直接的基因片段,如同DNA复制中的半保留复制一样,新合成的基因片段与模板链形成新的DNA双链,经反复的变性(denature)引物退火(anerling)和引物延伸(extention)三步循环,前一循环合成的DNA链成为下一循环引物结合的模板,每循环一次,反应体系的DNA的量就增加一倍,20-30次反复循环,即可由微量的DNA模板开始获得大量的DNA特异片段。近年来,PCR迅速发展,已深入生命科学的各个领域,技术方法不端完善,基本的PCR实验技术主要有经典PCR技术、RT-PCR技术、免疫-PCR技术、PCR-SSCP技术等。pcr检测做不好都有哪些原因?江西推荐pcr
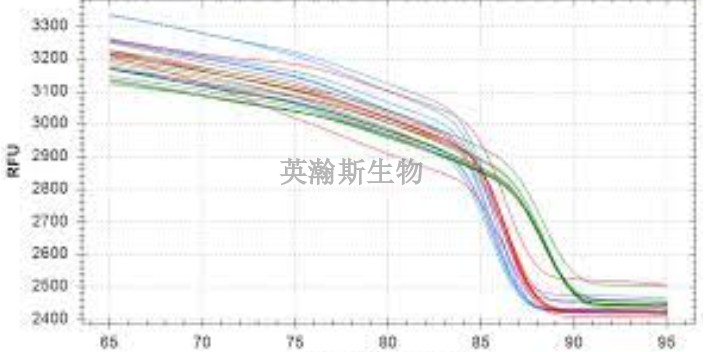
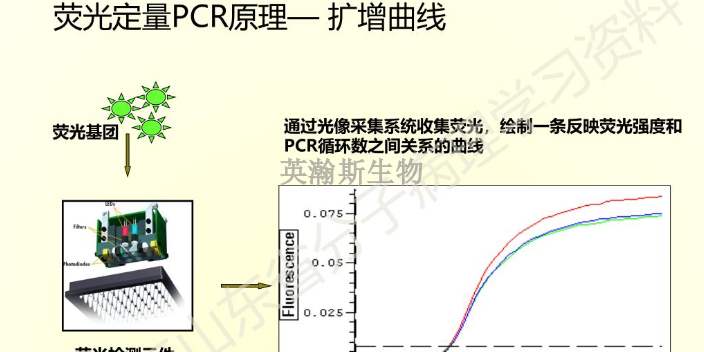
PCR实验技术中的RT-PCR一步法和两步法的优缺点介绍:一步法:利用同一缓冲液,在同一体系中加入逆转录酶、引物、Taq酶、4种dNTP直接进行mRNA反转录与PCR扩增。发现Taq酶不*具有DNA多聚酶的作用,而且具有反转录酶活性,可利用其双重作用在同一体系中直接以mRNA为模板进行反转录和其后PCR扩增,从而使mRNA的PCR步骤更为简化。所需样品量减少到比较低限度,临床小样品的检测非常有利。用一步法扩增可检测出总RNA中小于1ng的低丰度mRNA.该法可用于低丰度mRNA的cDNA文库的构建及特异cDNA的克隆,并有可能与Taq酶的测序技术相组合,使得自动反转录、基因扩增与基因转录产物的测序在单管中进行。两步法:由于单管反应时RT和PCR都不能在比较好条件下进行并且容易相互干扰,常只适宜用基因特异引物扩增较短的基因,及定量PCR.两步法则是将RT和PCR分别进行,这样使得两个反应充分发挥各自的特点,更为灵活而且严谨,适合那些GC含量、二级结构严重的模板或者是未知模板,以及多个基因的RT-PCR。河北高质量pcr实验荧光定量pcr的ct值怎么分析?
PCR反应的比较大特点是具有较大扩增能力与极高的灵敏性,但令人头疼的问题是易污染,极其微量的污染即可造成假阳性的产生。主要原因有:1、标本间交叉污染;2、PCR试剂的污染;3、PCR扩增产物污染;4、实验室中克隆质粒的污染。一个好的实验室,要时刻注意污染的监测,考虑有无污染是什么原因造成的污染,以便采取措施,防止和消除污染。1、阳性对照:在建立PCR反应实验室及一般的检验单位都应设有PCR阳性对照,它是PCR反应是否成功、产物条带位置及大小是否合乎理论要求的一个重要的参考标志。2、阴性对照:每次PCR实验务必做阴性对照。它包括:(1)标本对照:被检的标本是血清就用鉴定后的正常血清作对照;被检的标本是组织细胞就用相应的组织细胞作对照。(2)试剂对照:在PCR试剂中不加模板DNA或RNA,进行PCR扩增,以监测试剂是否污染。3、重复性试验。4、选择不同区域的引物进行PCR扩增。
PCR是一种具有选择性的体外扩增DNA的片段的方法。其特异性由两个人工合成的引物DNA序列决定。所谓引物是指与待扩增核酸片段两端互补的寡核苷酸,其本质是ssDNA(单链DNA)的片段。指在引物指导下由酶催化的对特定模板(克隆或基因组DNA)的扩增反应,是模拟体内DNA复制过程,在体外特异性扩增基因片段的一种技术,在分子生物学中有普遍的应用,包括用于DNA作图、DNA测序、分子系统遗传学等。CR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解旋成单链,在DNA聚合酶的参与下,根据碱基互补配对原则复制成同样的两分子拷贝。在实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,加入设计引物,DNA聚合酶、dNTP就可以完成特定基因的体外复制。英瀚斯生物,专业分子检测平台,高质量pcr检测。
PCR出现非特异性扩增带的原因分析:PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。其次,是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶的量过多有时也会出现非特异性扩增。其对策有:①必要时,重新设计引物。②减低酶的量或调换另一来源的酶。③降低引物量,适当增加模板量,减少循环次数。④适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸,也叫两步法)。靠谱的pcr检测外包公司推荐!贵州什么是pcr实验室
英瀚斯生物,分子平台实验人员十年以上经验,专业pcr检测。江西推荐pcr
PCR实验技术中cDNA第二链的合成方法有以下几种:1、自身引导法合成的单链cDNA3'端能够形成一短的发夹结构,这就为第二链的合成提供了现成的引物,当***链合成反应产物的DNA:RNA杂交链变性后利用大肠杆菌DNA聚合酶ⅠKlenow片段或反转录酶合成cDNA第二链,***用对单链特异性的S1核酸酶消化该环,即可进一步克隆。但自身引导合成法较难控制反应,而且用S1核酸酶切割发夹结构时无一例外地将导致对应于mRNA5'端序列出现缺失和重排,因而该方法目前很少使用。2、置换合成法该方法利用链在反转录酶作用下产生的cDNA:mRNA杂交链不用碱变性,而是在dNTP存在下,利用RNA酶H在杂交链的mRNA链上造成切口和缺口。从而产生一系列RNA引物,使之成为合成第二链的引物,在大肠杆菌DNA聚合酶Ⅰ的作用下合成第二链。该反应有3个主要优点:(1)非常有效;(2)直接利用链反应产物,无须进一步处理和纯化;(3)不必使用S1核酸酶来切割双链cDNA中的单链发夹环。江西推荐pcr
南京英瀚斯生物科技有限公司致力于医药健康,以科技创新实现***管理的追求。公司自创立以来,投身于实验外包,动物模型构建,细胞分子实验,病理检测,是医药健康的主力军。英瀚斯生物始终以本分踏实的精神和必胜的信念,影响并带动团队取得成功。英瀚斯生物始终关注医药健康行业。满足市场需求,提高产品价值,是我们前行的力量。