- 品牌
- 司鼎;OriCell
聚合酶链式反应PCR技术的基本原理类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性-退火-延伸三个基本反应步骤构成:模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;引物的延伸:DNA模板-引物结合物在72℃、DNA聚合酶(如TaqDNA聚合酶)的作用下,以dNTP为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环变性-退火-延伸三过程就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。聚合酶链反应的试剂应分配到一次性的等分试样中。深圳实时PCR检测技术原理
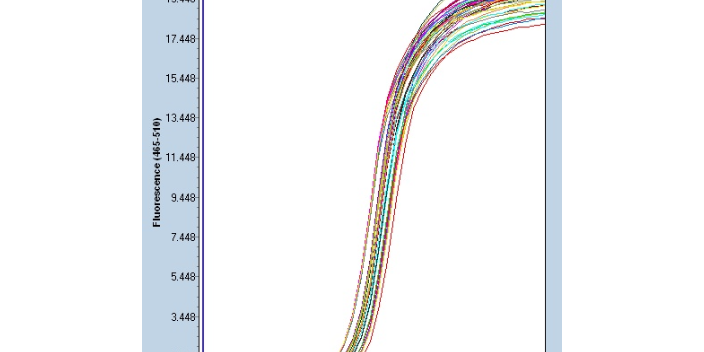
聚合酶链反应注意事项:在实验过程中要防止RNA的降解,保持RNA的完整性。在总RNA的提取过程中,注意避免mRNA的断裂;为了防止非特异性扩增,必须设阴性对照;内参的设定:主要为了用于靶RNA的定量。常用的内参有G3PD(甘油醛-3-磷酸脱氢酶)、β-Actin(β-肌动蛋白)等。其目的在于避免RNA定量误差、加样误差以及各PCR反应体系中扩增效率不均一各孔间的温度差等所造成的误差;PCR不能进入平台期,出现平台效应与所扩增的目的基因的长度、序列、二级结构以及目标DNA起始的数量有关。故对于每一个目标序列出现平台效应的循环数,均应通过单独实验来确定;防止DNA的污染: 采用DNA酶处理RNA; 在可能的情况下,将PCR引物置于基因的不同外显子,以消除基因和mRNA的共线性。徐州微量荧光定量PCR研究方案PCR反应的特异性决定因素为:引物与模板DNA特异正确的结合。

逆转录-聚合酶链反应的原理是:提取组织或细胞中的总RNA,以其中的mRNA作为模板,采用Oligo(dT)或随机引物利用逆转录酶反转录成cDNA。再以cDNA为模板进行PCR扩增,而获得目的基因或检测基因表达。完整RNA 的提取和纯化,是进行RNA 方面的研究工作,如Nothern杂交、mRNA 分离、RT-PCR、定量PCR、cDNA合成及体外翻译等的前提。所有RNA的提取过程中都有五个关键点,即样品细胞或组织的有效破碎;有效地使白复合体变性;对内源RNA酶的有效抑制;有效地将RNA从DNA和蛋白混合物中分离;对于多糖含量高的样品还牵涉到多糖杂质的有效除去。但其中很关键的是抑制RNA酶活性。RNA 的提取目前阶段主要可采用两种途径,提取总核酸,再用氯化锂将RNA 沉淀出来;直接在酸性条件下抽提,酸性下DNA与蛋白质进入有机相而RNA 留在水相。种提取方法将导致小分子量RNA 的丢失,目前该方法的使用频率已很低。
聚合酶链式反应:三步:变性:模板DNA加热变性;复性,引物与它的靶序列发生退火,引物DNA量多,使引物和模板在局部形成杂交链;延伸,4种dNTP 与 Mg2+ 存在的条件下,DNA合成酶催化以引物为起始点,按5'-3'方向进行延伸。上面三步为一个循环,可使拷贝数到达2*E-6-2*E-7。PCR产物一段双链DNA,是由它的末端引物的5’端决定的。首轮扩增,其产物是大小不均一的DNA分子。长度应大于两引物之间的长度。在第二个循环中,由于5'固定,其产物长度就由等于两引物之间的长度。所以几十个循环后就会产生大量的两引物之间序列。聚合酶链式反应准备:引物内部不应出现互补序列。

聚合酶链反应的常见问题分析与解决方法:扩增产物出现多条带(杂带):引物用量偏大,引物的特异性不高。应调换引物或降低引物的使用量。循环的次数过多。适当增加模板的量,减少循环次数。酶的用量偏高或酶的质量不好,应降低酶量或调换另一来源的酶。退火温度偏低,退火及延伸时间偏长。应提高退火温度,减少变性与延伸时间,也可采用二种温度的PCR扩增。以2度为梯度设计梯度PCR反应优化退火温度。 样品处理不当。Mg2+浓度偏高,因适当调整Mg2+使用浓度。若为PCR试剂盒,也可能时试剂盒本身质量有问题。复制提前终止。使用非热启动的聚合酶时常有发生。冰上准备反应体系或采用热启动聚合酶。许多疾病的未知病因的测序正在通过聚合酶链反应得到解决。常州实时Real-time PCR应用
聚合酶链式反应的特异性依赖于与靶序列两端互补的寡核苷酸引物。深圳实时PCR检测技术原理
聚合酶链式反应:定量PCR允许实时定量和检测特定的DNA序列,因为它在合成过程中测量浓度。有两种方法可以同时检测和定量。种方法是使用在DNA双链之间非特异性保留的荧光染料。第二种方法涉及编码特定序列并被荧光标记的探针。只有在探针与其互补DNA杂交后,才能使用这些方法检测DNA。一种有趣的技术组合是实时PCR和逆转录。这种复杂的技术被称为RT-qPCR,允许定量少量RNA。通过这种组合技术,mRNA被转化为cDNA,并用qPCR进一步定量。这种技术降低了聚合酶链反应终点出错的可能性,增加了检测与遗传疾病如病症相关的基因的机会。实验室使用RT-qPCR来灵敏地测量基因调控。深圳实时PCR检测技术原理
上海司鼎生物科技有限公司是一家有着雄厚实力背景、信誉可靠、励精图治、展望未来、有梦想有目标,有组织有体系的公司,坚持于带领员工在未来的道路上大放光明,携手共画蓝图,在上海市等地区的医药健康行业中积累了大批忠诚的客户粉丝源,也收获了良好的用户口碑,为公司的发展奠定的良好的行业基础,也希望未来公司能成为*****,努力为行业领域的发展奉献出自己的一份力量,我们相信精益求精的工作态度和不断的完善创新理念以及自强不息,斗志昂扬的的企业精神将**上海司鼎生物科技供应和您一起携手步入辉煌,共创佳绩,一直以来,公司贯彻执行科学管理、创新发展、诚实守信的方针,员工精诚努力,协同奋取,以品质、服务来赢得市场,我们一直在路上!

Real-time PCR-传统定量PCR:1)内参照法:在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板。2)竞争法:选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。PCR反应的很大特点是具有较大扩增能力与极高的灵敏性。深圳微量荧光PCR设计公司Real-time PCR原理:基于探针...
- 珠海实时荧光定量PCR技术服务 2023-08-11
- 温州组织荧光PCR原理及步骤 2023-08-11
- 上海微量RT-PCR检测技术供应商 2023-08-11
- 珠海骨头Real-time PCR原理 2023-08-10
- 武汉血液数字PCR技术服务 2023-08-10
- 连云港血液数字PCR原理及步骤 2023-08-10
- 宁波骨头定量PCR应用 2023-08-09
- 武汉血液PCR检测技术设计公司 2023-08-09
- 苏州细胞Real-time PCR服务 2023-08-09
- 徐州实时PCR检测技术服务 2023-08-09



- 杭州细胞Real-time PCR原理及步骤 2023-08-08
- 上海特殊样本数字PCR哪家好 2023-08-07
- 常州特殊样本PCR检测技术哪家好 2023-08-07
- 珠海特殊样本定量PCR原理 2023-08-07
- 徐州实时Real-time PCR供应商 2023-08-07
- 上海血液荧光定量PCR原理 2023-08-07




- 低毒性线性PEI转染试剂储存条件 07-14
- 深圳COS-7线性PEI转染试剂储存条件 07-14
- 广州HEK-293线性PEI转染试剂 07-14
- 高效线性PEI转染试剂作用 07-14
- 合肥细胞实验线性PEI转染试剂使用方法 07-14
- 深圳高效线性PEI转染试剂注意事项 07-14
- 南京无菌线性PEI转染试剂使用方法 07-13
- 上海HEK-293线性PEI转染试剂现货 07-13
- 合肥线性PEI转染试剂是干什么的 07-13
- 杭州高效转染线性PEI转染试剂储存条件 07-13