- 品牌
- 司鼎;OriCell
聚合酶链式反应是80年代中期发展起来的体外核酸扩增技术。工具/原料:扩增缓冲液,四种dNTP,引物,模板DNA,Taq DNA聚合酶, Mg离子,蒸馏水。模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA 链互补的半保留复制链。重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。聚合酶链式反应准备:PCR所用的酶主要有两种来源:Taq和Pfu,分别来自两种不同的噬热菌。无锡特殊样本荧光定量PCR设计公司

聚合酶链反应允许快速生产短DN段,即使已知的引物序列不超过两个。聚合酶链反应的这种能力增强了许多方法,例如生成杂交 探针用于 Southern 或northern blot 杂交。聚合酶链反应为这些技术提供了大量的纯DNA,有时是单链的,甚至可以从非常少量的起始材料中进行分析。脱氧核糖核酸测序的任务也可以通过聚合酶链反应来辅助完成。已知的DN段可以很容易地从患有遗传疾病突变的患者体内产生。对扩增技术的修饰可以从完全未知的基因组中提取片段,或者可以产生感兴趣区域的单链。聚合酶链反应有许多应用于传统的 DNA克隆。它可以从更大的基因组中提取片段以插入载体,这可能只有少量可用。使用一组“载体引物”,它还可以分析或提取已经插入载体的片段。聚合酶链反应方案的一些改变可以产生突变(通用的或定点的)插入片段。宁波组织RT-PCR检测技术原理聚合酶链式反应的模板的取材主要依据PCR的扩增对象,可以是病原体标本如病毒、细菌、菌类等。
聚合酶链式反应准备:引物内部不应出现互补序列。两个引物之间不应存在互补序列,尤其是避免3 ′端的互补重叠。引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增。引物3‘端的碱基,特别是很末及倒数第二个碱基,应严格要求配对,很好选择是G和C。引物的5′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等。
聚合酶链反应的常见问题分析与解决方法:无扩增条带:酶失活或在反应体系中未加入酶。Taq DNA聚合酶因保存或运输不当而失活,往往通过更换新酶或用另一来源的酶以获得满意的结果。模板含有杂质。特别是对甲醛固定及石蜡包埋的组织常含甲酸,造成DNA脱嘌呤而影响PCR的结果。变性温度是否准确:PCR仪指示温度与实际温度是否相符,过高酶在前几个循环就迅速失活;过低则模板变性不彻底。反应系统中污染了蛋白酶及核酸酶,应在未加Taq酶以前,将反应体系95℃加热5-10 min。引物变质失效。人工合成的引物是否正确。是否纯化,或因储存条件不当而失活。引物错误。利用BLAST检查引物特异性或重新设计引物。 DNA凝胶电泳时加入阳性对照,确保不是DNA凝胶和PCR程序的问题。重叠延伸聚合酶链反应可以将缺失、插入或点突变引入DNA序列。
聚合酶链式反应的常见问题:靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样内或溅出离心管外。除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管及样进头等均应一次性使用。必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。聚合酶链反应具有定量合成产物的优点。无锡特殊样本荧光定量PCR设计公司
嵌套聚合酶链反应:通过减少DNA非特异性扩增的背景,提高DNA扩增的特异性。无锡特殊样本荧光定量PCR设计公司
聚合酶链式反应准备:PCR引物设计:PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。引物设计的基本原则:引物长度:15-30bp,常用为20bp左右。引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C 过多易出现非特异条带。ATGC很好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列参照。无锡特殊样本荧光定量PCR设计公司

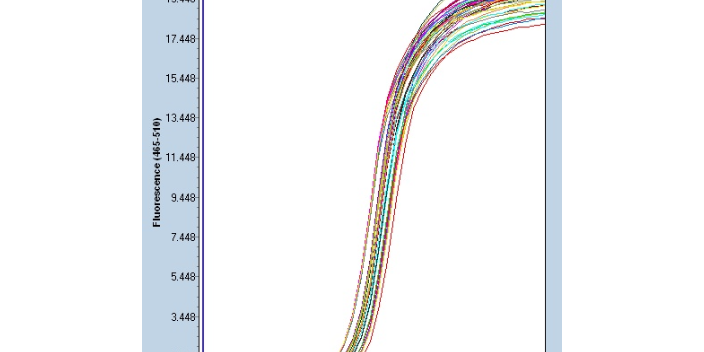
Real-time PCR-传统定量PCR:1)内参照法:在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板。2)竞争法:选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。PCR反应的很大特点是具有较大扩增能力与极高的灵敏性。深圳微量荧光PCR设计公司Real-time PCR原理:基于探针...
- 珠海实时荧光定量PCR技术服务 2023-08-11
- 温州组织荧光PCR原理及步骤 2023-08-11
- 上海微量RT-PCR检测技术供应商 2023-08-11
- 珠海骨头Real-time PCR原理 2023-08-10
- 武汉血液数字PCR技术服务 2023-08-10
- 连云港血液数字PCR原理及步骤 2023-08-10
- 宁波骨头定量PCR应用 2023-08-09
- 武汉血液PCR检测技术设计公司 2023-08-09
- 苏州细胞Real-time PCR服务 2023-08-09
- 徐州实时PCR检测技术服务 2023-08-09


- 杭州细胞Real-time PCR原理及步骤 2023-08-08
- 上海特殊样本数字PCR哪家好 2023-08-07
- 常州特殊样本PCR检测技术哪家好 2023-08-07
- 珠海特殊样本定量PCR原理 2023-08-07
- 徐州实时Real-time PCR供应商 2023-08-07
- 上海血液荧光定量PCR原理 2023-08-07




- 无锡HEK-293线性PEI转染试剂原理 07-11
- 南通线性PEI转染试剂价格 07-11
- 上海高表达线性PEI转染试剂注意事项 07-11
- 杭州高纯度线性PEI转染试剂原理 07-11
- 深圳40000Da线性PEI转染试剂哪家好 07-11
- 南京HEK-293线性PEI转染试剂供应商 07-11
- 武汉高效线性PEI转染试剂注意事项 07-11
- 苏州40000Da线性PEI转染试剂效果好吗 07-11
- 广州线性PEI转染试剂厂家推荐 07-10
- 杭州动物细胞线性PEI转染试剂注意事项 07-10